
雷特综合征(Rett syndrome)是一种严重的神经发育障碍,主要是由基因MeCP2(methyl-CpG-binding protein 2, 甲基CpG结合蛋白2)突变引起。最初,MeCP2被认为是一种重要的脑蛋白,它通过它的甲基结合结构域(MBD)与甲基化的CpG(mCG)结合,起到转录抑制的作用。然而,在早期大脑发育过程中,MeCP2的产后积累与全基因组范围内羟甲基化胞嘧啶(hmC)和甲基化CpA(mCA)的高水平积累同时发生,这表明MeCP2也可能识别并结合含有这些修饰核苷酸的DNA序列。MeCP2同时识别mCA、hmC和mCG的能力导致了关于它在转录调控中的功能的相互矛盾的结论,因为这些胞嘧啶修饰与转录的抑制(mCA和mCG)或激活(hmC)有关。明确确定MeCP2起作用的靶序列将有助于澄清这一问题。
CA重复序列(CAn)占小鼠基因组的约1%,属于微卫星家族。它们广泛分布于整个基因组,并被证明会影响附近基因的转录。法国研究人员的最近数据显示,CAn在多种细胞类型中发生甲基化(mCAn)或羟甲基化(hmCAn)。在寻找能特异性识别和结合这些CA重复序列的蛋白时,他们发现MeCP2可特异性读取CA重复序列。
基于此,他们在一项新的研究中,猜测CAn的甲基化状态对MeCP2的识别和结合至关重要,可能是通过识别CA重复序列中具有不同亲和力的修饰核苷酸,这与它在转录调节中的神经元功能有关。相关研究结果发表在2021年6月25日的Science期刊上,论文标题为“MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion”。
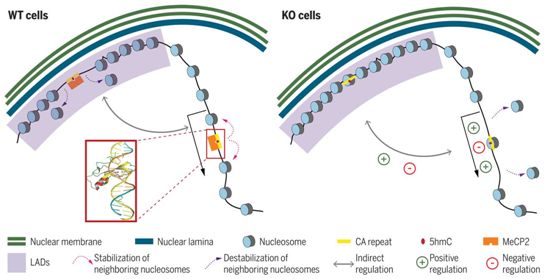
这些作者发现,在MBD家族中,MeCP2是唯一能特异性识别和结合CA重复序列的蛋白质,其亲和力比mCG和mCA强得多。MeCP2以链特异性的方式选择性地识别CA重复序列,并且需要至少五个连续的CA二核苷酸才能最佳地与DNA结合。虽然MeCP2能在体外与修饰的和未修饰的CA重复序列结合,但它对羟甲基化的CA重复序列表现出令人印象深刻的选择性,这些羟甲基化的CA重复序列是DNA(胞嘧啶-5)-甲基转移酶3A修饰CA重复序列而产生的。这种修饰的胞嘧啶只有在位于CA重复序列内时,才会成为MeCP2在这种重复序列周围聚集和扩散的成核点,而这又与核小体排斥有关。此外,MeCP2的缺失导致核纤层相关结构域(lamina-associated domain, LAD)内的核小体密度广泛增加以及位于LAD外面的富含CA重复序列的基因的转录失调。
这些作者还通过解析出MeCP2与hmCAn结合在一起时的晶体结构,剖析了MeCP2识别羟甲基化CA重复序列的分子基础。这种羟甲基化CA重复序列形成了一种明确确定的DNA形状,其几何形状有很大的改变,包括一个加宽的主槽和负卷参数,精确地位于修饰位点。他们发现,羟甲基化CA重复序列的分子识别是通过Arg133发生的,Arg133是MeCP2的一个关键氨基酸残基,该位点的突变可导致雷特综合征。
综上所述,这项研究鉴定出MeCP2是羟甲基化CA重复序列的DNA结合蛋白,靶向富含5hmC-CA的DNA序列,这些DNA序列特异地位于一条链上。这些作者的数据在分子水平上为雷特综合征的起源提供了新见解,并表明这种神经发育障碍可能被看作是一种染色质疾病,源于发生突变的MeCP2无法结合CA重复序列和保护这种重复序列免受核小体入侵。这些研究结果开辟了一个以前没有探索过的研究领域,重点是了解与微卫星和其他重复序列结合的特定蛋白在未知病因的神经系统疾病中的作用。








